
¿Sabías que en el interior de tus células se encuentra el libro más antiguo y mejor conservado de la historia del ser humano? Comprendiendo el ADN, es posible entender el pasado. Hace cinco millones de años, una región del cromosoma cinco se duplicó en el ADN de un ancestro que compartimos con el chimpancé. El análisis de las secuencias compartidas entre humanos y chimpancés nos permite entender qué cambios han ido surgiendo a lo largo de la historia. Resulta casi inverosímil pensar que los mayores actos del ser humano de hoy en día, por relevantes y extraordinarios que parezcan actualmente, caerán en el olvido más pronto que tarde. Mientras tanto, las minúsculas modificaciones que están ocurriendo en nuestro genoma pasarán a la historia, y es probablemente la información más valiosa que podrá recibir la población que habitará la tierra dentro de cinco millones de años.
La Atrofia Muscular Espinal
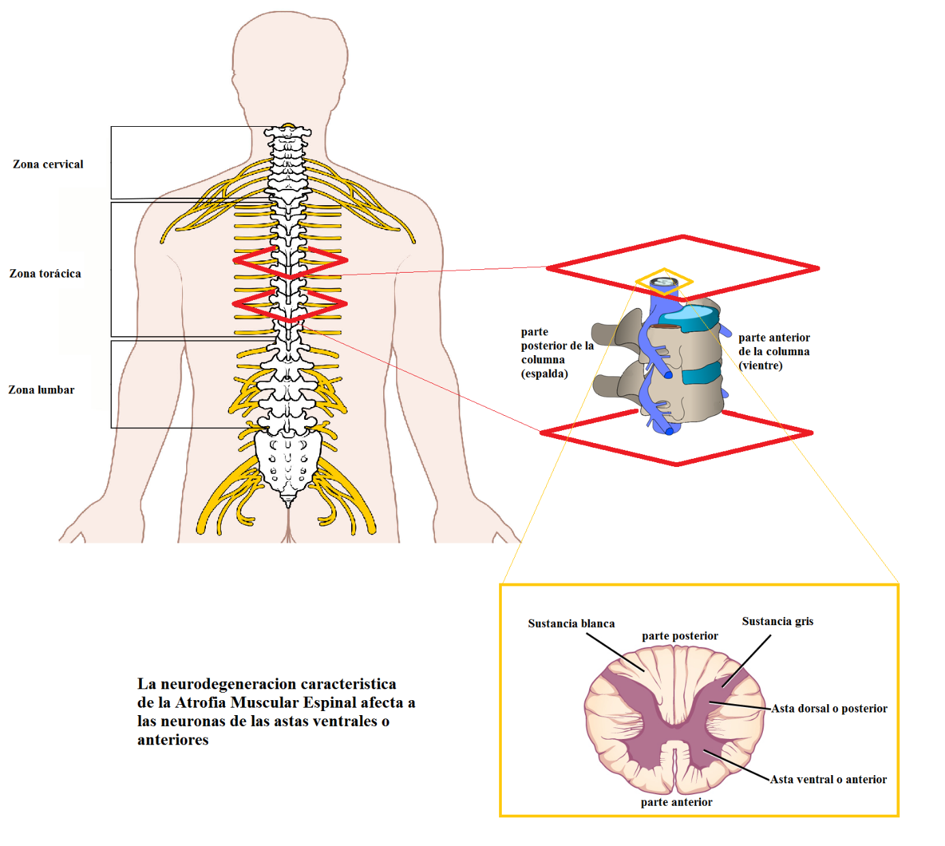
La Atrofia Muscular Espinal (AME) es una enfermedad neurodegenerativa que afecta principalmente a las neuronas de la asta anterior de la medula espinal. Se trata de la primera causa de mortalidad infantil por enfermedad hereditaria. Sus manifestaciones varían en función del tipo clínico de manifestación (tipos I, II, III y IV, de más severo a menos), pero por lo general son las funciones motoras las principales afectadas, siendo la debilidad y la atrofia muscular los síntomas más comunes. La neurodegeneración está asociada a un problema genético relacionado con la proteína SMN.
Entiende qué es el ARN y la importancia de su metabolismo
El objetivo del presente artículo es discutir sobre la patogénesis de la AME. Es decir, de qué manera las mutaciones del gen SMN1 dan lugar a la neurodegeneración y como consecuencia a la atrofia muscular. Antes de ello, es importante aclarar ciertos conceptos esenciales de la biología.
Todo el mundo está familiarizado con el ADN: la molécula que define quienes somos, la base de la herencia. Sin embargo, la molécula de ARN es por lo general mucho menos conocida, pero su rol es tan importante como el del ADN. El ARN es sintetizado a partir de una región de ADN que conocemos como gen. Para entenderlo, hay que imaginar que los genes son “mensajes celulares” que indican lo que hay que hacer. El ARN no es más que una transcripción de estos mensajes celulares de ADN a un idioma que la célula entiende. Una vez que la molécula de ARN es sintetizada, esta puede ejercer su función directamente, o puede dar lugar a otra molécula diferente: la proteína (roles estructurales o funcionales). Por supuesto, la realidad no se limita a estas dos etapas de transcripción sin/y traducción para explicar el funcionamiento de una célula. En efecto, hay subetapas igualmente importantes que permiten afinar los mensajes.
Profundizando a un nivel de detalle ligeramente superior, la transcripción de ciertos genes que codifican proteínas da lugar a una molécula inmadura de ARN conocida como ARN pre-mensajero (o ARN precursor). Hay que imaginar esta molécula de ARN pre-mensajero como un manual de instrucciones para la célula que contiene información útil (regiones del ARN conocidas como exones) e información inútil (regiones conocidas como intrones), separando los párrafos de información útil. La célula utiliza entonces un sistema de splicing o empalme del ARN, que consiste simplemente en eliminar los intrones para que el texto de exones sea legible. Una vez que el sistema de empalme ha ejercido su función sobre el ARN pre-mensajero, ésta madura y da lugar a la molécula de ARN mensajero, listo para ser enviado al citoplasma celular y set traducido en proteína para ejercer su función.
La biología de la Atrofia Muscular Espinal y el metabolismo del ARN
El ser humano posee dos proteínas llamadas SMN: SMN1 y SMN2. Estas dos macromoléculas difieren únicamente en un solo nucleótido (letras del ADN, que dan lugar a las letras del ARN, y que dan lugar a los bloques o ladrillos de las proteínas). Pero esta pequeña diferencia es suficiente para separar sus caminos dentro de la célula: SMN1 es perfectamente funcional, mientras que SMN2 es inestable y su función se ve muy reducida en el 90% de los casos. La mayoría de los pacientes de AME se caracterizan por poseer mutaciones (deleciones) en el gen que codifica la proteína SMN1 (concretamente, a nivel de los exones número 7 y 8: parte de las instrucciones útiles) pero poseen algunas copias de SMN2 funcionales (Arnold & Fischbeck, 2018). La severidad de la enfermedad disminuye con el aumento del porcentaje de SMN2 funcional (Lefebvre et al., 1997). Esto, entre otras razones, invita a pensar que la patología es el resultado de una deficiencia de SMN funcional, y no una ausencia total de la misma.
Hasta aquí no se ha dicho nada revolucionario: la proteína SMN1 es funcional, y su disminución correlaciona con la aparición de la AME. El lector se preguntará, en este momento, cuál es el rol de esta proteína, que resulta tan determinante para el devenir de la célula. La proteína SMN se expresa de manera ubiquitaria (en todos los tejidos), pero el nivel de expresión puede variar de un tejido a otro:

La proteína SMN se encuentra tanto en el citoplasma como en el núcleo celular, y cumple varias funciones, relacionadas en gran parte con el metabolismo del ARN. Cuando se encuentra en el núcleo, SMN forma complejos con otras proteínas, y está implicada en funciones como el empalme o splicing del ARN. Cuando se encuentra en el citoplasma, SMN se asocia con moléculas de ARNsn (small nuclear RNA, o pequeños ARN nucleares), dando lugar a unas estructuras complejas denominadas pequeñas ribonucleoproteínas nucleares. La estabilidad de estas estructuras depende del buen funcionamiento de los complejos de SMN nucleares mencionados más arriba. En otras palabras, SMN es una proteína implicada en la maduración del ARN mediante el proceso de empalme (Arnold & Fischbeck, 2018). La hipótesis principal para explicar la muerte neuronal es que la disminución de la proteína SMN1 funcional da lugar a problemas de maduración del ARN que resultan letales para las neuronas.
Una incógnita
Si SMN es una proteína ubiquitaria, ¿por qué la mutación afecta únicamente a las neuronas motoras, causando la atrofia muscular típica de la AME, y el resto de los tejidos se ven inafectados? Una posibilidad es que existan varios complejos de empalme diferentes en cada tejido, y que cada uno esté especializado en la regulación de genes diferentes. Siguiendo este razonamiento, podría plantearse la hipótesis de que los genes regulados por SMN en las neuronas motoras son esenciales para la supervivencia de la célula (por ejemplo, un gen que regula el ciclo celular), lo que explicaría una neurodegeneración en un contexto de mutación. En la misma línea, podría plantearse que los genes regulados por este mismo complejo en el resto de los tejidos no son esenciales para la supervivencia celular (o existen genes redundantes, es decir, que cumplen la misma función que los genes afectados, compensando el defecto) y que existen otros complejos de empalme funcionales que permiten el buen funcionamiento del metabolismo del ARN.
Para responder a esta pregunta, algunos estudios han realizado estudios de transcriptómica, con el objetivo de identificar los genes regulados por SMN. En el estudio de Maeda et al. (2014) se utilizó un modelo animal de la Atrofia Muscular Espinal: un ratón modificado genéticamente para imitar la mutación SMN1 humana. En este estudio se estudió el transcriptoma (identificación y cuantificación de los ARN mensajeros, productos de los diferentes genes) de las neuronas motoras de los ratones. Gracias a este estudio, se identificaron 3094 genes con una mayor tasa de transcripción en ratones con la mutación SMN típica de la Atrofia Muscular Espinal. Es decir, la proteína SMN, en condiciones normales, podría estar asociada a la inhibición de la sobreexpresión de estos más de 3000 genes para mantenerlos dentro de unos niveles normales. La disfunción de SMN tendría como consecuencia una sobreexpresión de estos. Por otra parte, en este mismo estudio fueron identificados 6964 genes infraexpresados en ratones con la mutación de SMN. En cuanto a éstos, podría tratarse de genes regulados positivamente por la proteína SMN funcional.
Entre los 3094 genes sobreexpresados en las neuronas motoras de los ratones modelos de la AME, se encontraron genes relacionados con pluripotencia celular (capacidad de diferenciación celular importante) y con la proliferación celular. Entre los 6964 genes infraexpresados se encontraron genes relacionados con el desarrollo y la actividad de las neuronas. Estos resultados sugieren que la deficiencia de la proteína SMN afecta al desarrollo neuronal y al mantenimiento de las neuronas motoras. Esto es coherente con la hipótesis de que los genes esenciales de cada tejido podrían ser regulados por diferentes complejos de empalme, y que la mutación de SMN no tendría por qué afectar al resto de tejidos. No obstante, para analizar más a fondo esta hipótesis, sería necesario estudiar el transcriptoma de otros tejidos utilizando el mismo modelo de Atrofia Muscular Espinal. Por el momento, se desconoce por qué la disfunción de SMN en el metabolismo del ARN afecta preferencialmente a la neurona motora (Arnold & Fischbeck, 2018).
Tratamiento de la Atrofia Muscular Espinal
El estado actual de la cura ha mejorado considerablemente con respecto a los últimos años, en los que lo máximo que se podía hacer por los pacientes era acompañarlos y ofrecerles ayuda en la realización de tareas cotidianas como la alimentación. Actualmente, la investigación ha avanzado siguiendo diferentes líneas para desarrollar una cura para la enfermedad, tomando como diana el origen genético de la misma.
Una de las vías que se han investigado para tratar la AME es la farmacológica, cuyo objetivo es aumentar la expresión de SMN2, de tal forma que la carencia de SMN1 sea compensada. Una de las muchas estrategias que se han empleado consiste en inhibir unas proteínas llamadas histonas deacetilasas (HDAC), implicadas en la regulación negativa de la expresión genética. Estas proteínas funcionan eliminando las acetilaciones del ADN. La acetilación del ADN es una modificación genética que lo mantiene en un estado de condensación reducida compatible con la expresión genética, por lo que la eliminación de la acetilación por parte de las HDAC estimula la compactación del ADN reduciendo así la genética. En resumen, la terapia consiste en inhibir un represor de la expresión genética, lo que resulta en un aumento de esta. Algunos ejemplos de fármacos inhibidores de HDAC son el butirato de sodio, el fenilbutirato de sodio y el ácido valproico. Otra estrategia farmacéutica consiste en la inhibición del proteasoma mediante fármacos como el bortezomib. El proteasoma es un complejo celular encargado de degradar las proteínas, por lo que el principio de esta estrategia es el mismo que para la anterior: la inhibición del proteasoma resulta en un aumento de la cantidad de proteínas. Las consideraciones que hay que tener en cuenta en estas dos estrategias son similares: hasta qué punto son vías específicas para aumentar la SMN2 (en otras palabras, ¿hay otras proteínas cuya expresión aumenta? ¿cuáles?). Por el momento, los resultados de los ensayos clínicos son prometedores, pero no definitivos. (Arnold & Fischbeck, 2018).
Paralelamente a las estrategias farmacológicas han surgido otras consistente en la sustitución genética. La idea de estas terapias consiste en introducir, por medio de un vector viral capaz de atravesar la barrera hematoencefálica, el gen SMN. Algunos ensayos realizados en ratones (modelos de AME) han mostrado resultados positivos, con un aumento considerable de la esperanza de vida: 250 días tras la terapia viral frente a 15,5 días sin tratamiento.
Por último, se han desarrollado algunas estrategias de remplazo celular. Estas terapias consisten en introducir células madre en los tejidos afectados, de manera que estas se diferencien en neuronas motoras sanas. Este enfoque ha sido muy poco explorado en el contexto de la AME.
Lo que ahora sabes (y lo que no)
El ARN juega un papel vital en la célula: es el precursor de las proteínas, unidades estructurales y funcionales de nuestras células. El metabolismo del ARN incluye etapas como la maduración de los precursores de ARN mensajero. Sin la maduración del ARN mensajero, la proteína resultante puede dejar de ser viable y esto puede tener consecuencias para la viabilidad de las células. Es el caso de la enfermedad neurodegenerativa conocida como Atrofia Muscular Espinal. Esta patología se caracteriza por una mutación genética de la proteína SMN1, implicada en la maduración del ARN. Sin esta proteína funcional, su homóloga SMN2 no es suficiente para restaurar las funciones celulares como el desarrollo y la proliferación de las células, en este caso las neuronas motoras. Aún resulta un misterio por qué la mutación genética tiene repercusiones únicamente a nivel de estas células nerviosas (y como consecuencias sobre los músculos inervados por estas, resultando en una atrofia muscular). Existen varias vías de desarrollo de tratamientos: terapia farmacológica, cuyo objetivo es aumentar de forma directa la cantidad de proteína SMN jugando sobre los reguladores genéticos de su expresión; la terapia genética, que busca la incorporación en la célula de la proteína SMN funcional mediante terapias virales; y la terapia celular, que propone la utilización de células madre para desarrollar nuevos tejidos funcionales. A pesar de que se ha avanzado considerablemente en la búsqueda de vías factibles, ninguna estrategia ha mostrado resultados definitivos.
Arnold, E. S., & Fischbeck, K. H. (2018). Spinal muscular atrophy. Neurogenetics, Part II, 148(38), 591–601. Fuente
Lefebvre, S., Burlet, P., Liu, Q., Bertrandy, S., Clermont, O., Munnich, A., … Melki, J. (1997). Correlation between severity and SMN protein level in spinal muscular atrophy. Nature Genetics, 16(3), 265–269. Fuente
Maeda, M., Harris, A. W., Kingham, B. F., Lumpkin, C. J., Opdenaker, L. M., McCahan, S. M., … Butchbach, M. E. R. (2014). Transcriptome Profiling of Spinal Muscular Atrophy Motor Neurons Derived from Mouse Embryonic Stem Cells. PLoS ONE, 9(9), 1–18. Fuente
Protein Atlas. (s.f.). SMN1 protein expression summary - The Human Protein Atlas. Recuperado 2 febrero, 2020. Fuente

